El Reglamento (UE) 2017/746 sobre productos sanitarios para diagnóstico in vitro (IVDR) ha supuesto un cambio estructural en la regulación de estos productos en Europa. Su objetivo es reforzar la seguridad, la trazabilidad y la evidencia clínica asociada a los dispositivos IVD, adaptando el marco normativo a las tecnologías emergentes y a las nuevas exigencias sanitarias.
En 2024, el Reglamento (UE) 2024/1860 ha introducido nuevas disposiciones transitorias para facilitar la transición hacia el IVDR sin comprometer la disponibilidad de productos esenciales. Este artículo analiza en detalle las actualizaciones normativas, los requisitos técnicos del IVDR y su impacto en fabricantes, laboratorios, distribuidores y profesionales sanitarios.Punto de partida. Requisitos clave
El nuevo marco normativo implica una transición profunda para todos los agentes económicos del sector: desde fabricantes hasta laboratorios clínicos. A continuación, se resumen los requisitos esenciales que definen el cumplimiento del IVDR, y que siguen plenamente vigentes para todos los productos sanitarios para diagnóstico in vitro comercializados en la Unión Europea.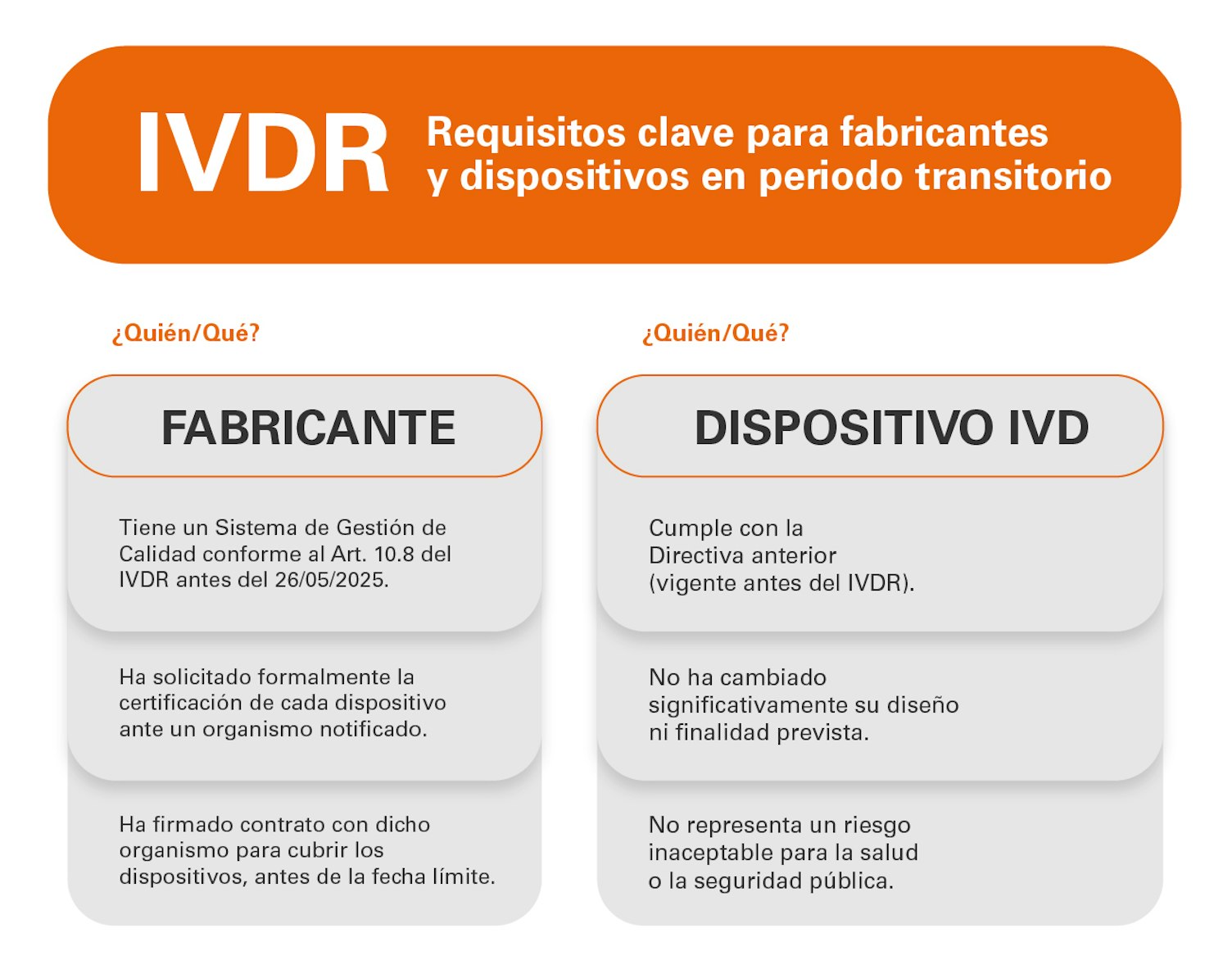
¿Qué es el IVDR?
El Reglamento (UE) 2017/746 sobre productos sanitarios para diagnóstico in vitro (IVDR) representa una transformación profunda del marco legal que regula estos productos en la Unión Europea. Sustituye a la anterior Directiva 98/79/CE (IVDD), vigente desde 1998, que había quedado desfasada frente a la evolución tecnológica, los nuevos modelos de atención sanitaria y las crecientes exigencias de seguridad, trazabilidad y transparencia del sector.
El IVDR fue diseñado para cerrar brechas regulatorias que la directiva anterior no abordaba con suficiente precisión. Entre ellas, la falta de control externo en la mayoría de los productos, la limitada exigencia de evidencia clínica, la escasa trazabilidad del producto una vez comercializado, o la ausencia de mecanismos robustos de vigilancia postcomercialización. Estas deficiencias se volvieron especialmente visibles en situaciones de crisis sanitaria y tecnológica, donde se requería una respuesta reguladora más exigente y coherente con los riesgos asociados.
La nueva regulación redefine el concepto de diagnóstico in vitro y amplía su alcance para incluir tecnologías emergentes como software, algoritmos predictivos, diagnósticos complementarios y dispositivos para autodiagnóstico. Además, establece un sistema de clasificación basado en el riesgo, lo que obliga a que la gran mayoría de los dispositivos sean ahora evaluados por un Organismo Notificado. Introduce también nuevos requisitos de evidencia científica, de funcionamiento analítico y clínico, además de exigir sistemas formales de gestión de calidad, trazabilidad mediante UDI (Identificación Única del Dispositivo) y vigilancia activa tras la comercialización.
El objetivo del IVDR no es solo incrementar la seguridad de los pacientes, sino también garantizar que los productos puestos en el mercado europeo cumplen con los estándares internacionales más altos de calidad, eficacia y transparencia. Para los fabricantes, esto implica un cambio de paradigma: ya no basta con demostrar cumplimiento técnico, sino que deben generar y mantener evidencia robusta y verificable a lo largo de todo el ciclo de vida del producto.
El IVDR busca profesionalizar el mercado del diagnóstico in vitro, generar mayor confianza en los dispositivos disponibles y reforzar la posición de Europa como mercado regulado de referencia. A cambio, plantea retos técnicos y organizativos que requieren una planificación sólida, recursos especializados y colaboración temprana con los Organismos Notificados.
Modificaciones recientes del IVDR: Reglamento (UE) 2024/1860
La Comisión Europea ha reconocido que los plazos originales para la plena aplicación del IVDR planteaban riesgos concretos de desabastecimiento en el mercado europeo, especialmente en el caso de productos críticos y dispositivos IVD con alta dependencia regulatoria. Por ello, mediante el Reglamento (UE) 2024/1860, se han ampliado los períodos transitorios bajo ciertas condiciones específicas.
La ampliación no es automática ni generalizada. Para acogerse a los nuevos plazos, los fabricantes deben cumplir requisitos como haber implementado un sistema de gestión de calidad (QMS), estar avanzando en la preparación de la documentación técnica conforme al IVDR, y no haber registrado incidentes graves que comprometan la seguridad del producto. Dependiendo de la clase de riesgo del dispositivo, los nuevos plazos para cumplir con el IVDR se extienden hasta 2027 o incluso 2029.
Esta prórroga selectiva busca asegurar la disponibilidad de productos sanitarios esenciales sin relajar los estándares regulatorios exigidos por el IVDR. Además, exige a los fabricantes mantener una planificación estricta y documentación demostrable de progreso hacia el cumplimiento.
Implementación gradual de EUDAMED
Uno de los pilares del IVDR es la creación de un entorno de datos transparente y accesible a través de la base de datos europea sobre productos sanitarios, EUDAMED. Aunque su lanzamiento completo ha sufrido retrasos, la Comisión Europea ha confirmado una implementación modular y gradual.
Actualmente, algunos módulos —como el de registro de actores económicos— ya están operativos. Otros, como los relacionados con vigilancia postcomercialización o estudios clínicos, estarán disponibles progresivamente, permitiendo que fabricantes y autoridades se adapten a sus funcionalidades de forma escalonada. Una vez esté completamente activa, EUDAMED será la herramienta central para el registro de productos, la gestión de incidencias, la trazabilidad mediante UDI y la consulta pública de información técnica.
La entrada en vigor plena de EUDAMED será también un hito en la armonización regulatoria dentro del mercado único europeo y reforzará la transparencia frente a pacientes, profesionales y autoridades.
Nuevas obligaciones de notificación ante interrupciones de suministro
Otra novedad crítica del Reglamento 2024/1860 es la obligación de notificar interrupciones de suministro de determinados dispositivos IVD considerados esenciales para la salud pública. Esta obligación recae sobre los fabricantes y distribuidores, quienes deberán informar con antelación suficiente a las autoridades competentes y a las instituciones sanitarias afectadas.
El objetivo es permitir una respuesta rápida por parte del sistema sanitario ante posibles escaseces, ya sea mediante alternativas clínicas, ajustes logísticos o medidas regulatorias excepcionales. Esta obligación refuerza la corresponsabilidad de los operadores económicos no solo en la calidad y seguridad del producto, sino también en la continuidad de su disponibilidad en el mercado.
Impacto del IVDR: ¿a quién se aplica y cómo afecta?
El IVDR afecta directamente a todos los fabricantes de productos sanitarios para diagnóstico in vitro que deseen comercializar sus dispositivos en la Unión Europea. Sin embargo, su alcance es mucho más amplio.
Los importadores y distribuidores también tienen nuevas responsabilidades, incluyendo el control documental, la verificación de marcado CE y la obligación de colaborar activamente con fabricantes y autoridades en caso de incidentes o retiros de producto.
Además, el reglamento tiene implicaciones importantes para los laboratorios clínicos e instituciones sanitarias que desarrollan o utilizan test de fabricación propia (LDT, por sus siglas en inglés). Estos solo podrán seguir utilizándose si cumplen estrictamente las condiciones del artículo 5.5 del IVDR, lo que incluye justificaciones clínicas específicas, documentación técnica equivalente a la de un fabricante y ausencia de alternativas comerciales viables. Por tanto, el IVDR obliga a todos los actores a elevar sus estándares de calidad, trazabilidad y documentación.
Conclusión: cumplimiento técnico, seguridad y continuidad del mercado
El IVDR representa un cambio de paradigma en la regulación de los dispositivos de diagnóstico in vitro en Europa. No se trata solo de cumplir con una nueva norma, sino de adaptarse a un modelo regulador basado en evidencia, trazabilidad y transparencia.
Las modificaciones introducidas en 2024 ofrecen margen adicional para que los operadores económicos completen la transición sin poner en riesgo la disponibilidad de productos críticos. Pero también refuerzan la necesidad de avanzar sin dilaciones, con sistemas robustos, documentación sólida y planificación estratégica.
Fabricantes, distribuidores, laboratorios y profesionales sanitarios deben comprender que el IVDR no es un mero requisito administrativo, sino una herramienta para garantizar la seguridad diagnóstica y la calidad asistencial en toda la UE. Adaptarse a él es, además de una obligación regulatoria, una inversión en confianza, sostenibilidad e innovación.
¿Cómo te puede ayudar SGS?
SGS, acreditada como organismo notificado para IVDR, ofrece un acompañamiento técnico y normativo integral a lo largo de todo el proceso. Entre sus servicios se incluyen la evaluación de conformidad y la revisión de expedientes técnicos, la realización de auditorías in situ y la verificación de sistemas de calidad, así como formación especializada sobre la aplicación del reglamento y la verificación del cumplimiento para productos “in-house”.
Este enfoque se apoya en un profundo conocimiento técnico del sector de los productos sanitarios, en una red internacional de expertos y en la experiencia acumulada en la implementación de los reglamentos MDR e IVDR.
Nuestro enfoque combina el conocimiento técnico en productos sanitarios con una red internacional y la experiencia acumulada en reglamentos MDR e IVDR.



